
EpiTect Hi-C Kit
Pour cartographie haute résolution du repliement de la chromatine, assemblage haute qualité des séquences génomiques, phasage des haplotypes et identification des réarrangements chromosomiques
Pour cartographie haute résolution du repliement de la chromatine, assemblage haute qualité des séquences génomiques, phasage des haplotypes et identification des réarrangements chromosomiques
✓ Traitement automatique des commandes en ligne 24 h/24 7 j/7
✓ Assistance technique et produits pertinente et professionnelle
✓ Commande (ou réapprovisionnement) rapide et fiable
N° de réf. / ID. 59971
✓ Traitement automatique des commandes en ligne 24 h/24 7 j/7
✓ Assistance technique et produits pertinente et professionnelle
✓ Commande (ou réapprovisionnement) rapide et fiable
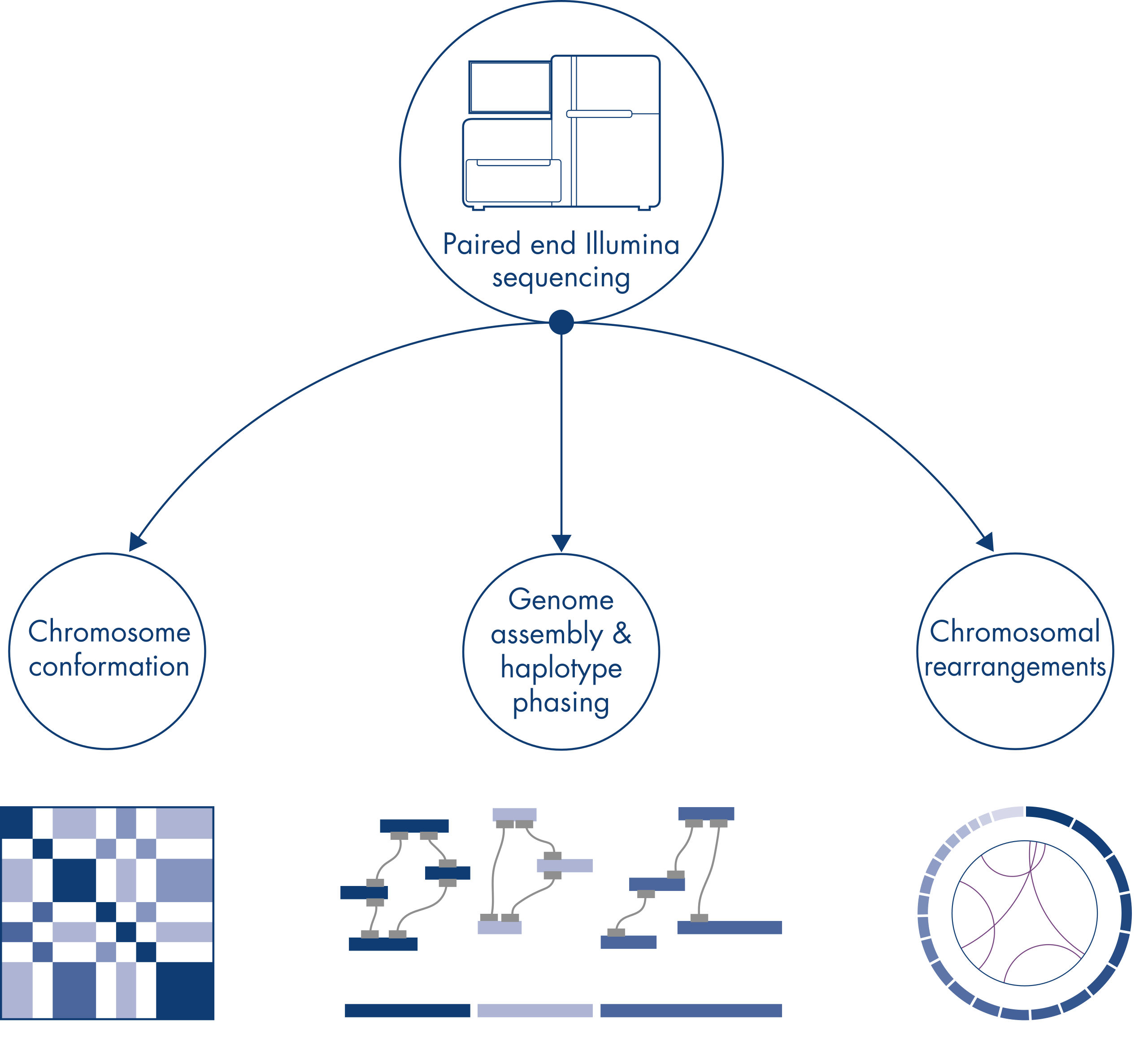
Hi-C a été conçu à l’origine comme une technique puissante de capture de la conformation des chromosomes à l’échelle du génome, qui permettait de caractériser le repliement de la chromatine à une résolution de l’ordre du kb. Toutefois, la technologie a également d’autres applications importantes. Par exemple, Hi-C est utilisé pour générer des ensembles de génomes très contigus, avec peu d’échafaudages très longs, à partir d’organismes sans génome de référence connu. En outre, Hi-C est également très utile pour le phasage des haplotypes et la détection des réarrangements chromosomiques.
L’EpiTect Hi-C Kit propose un protocole robuste, mais simple et rapide, avec de faibles exigences en matière d’intrants cellulaires, ce qui permet de générer des bibliothèques de NGS Illumina Hi-C de haute qualité à partir de cellules réticulées en moins de 2 jours.
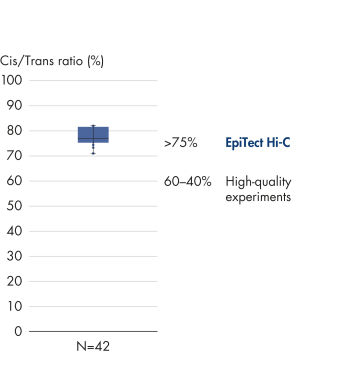
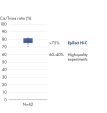
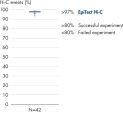
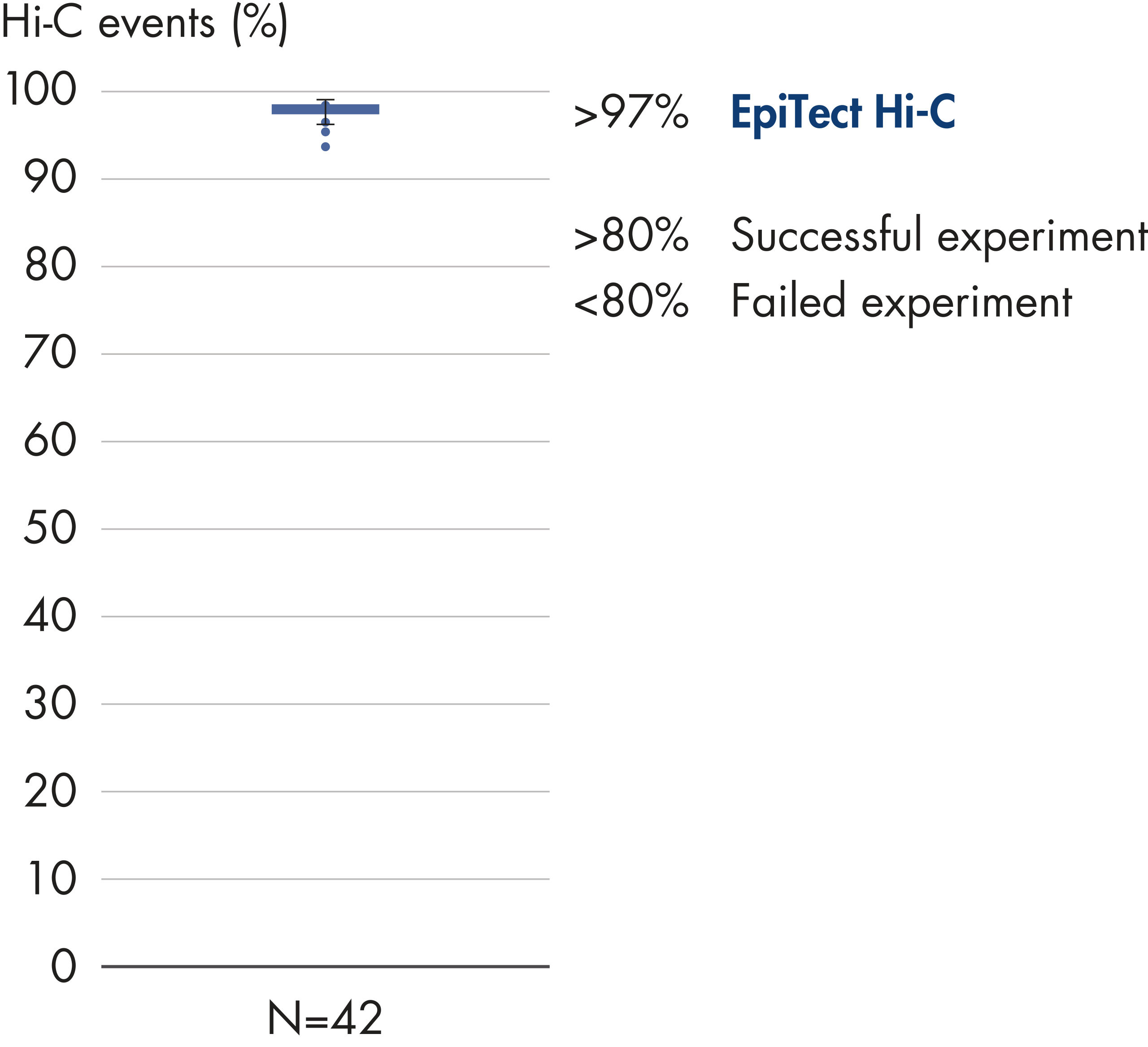
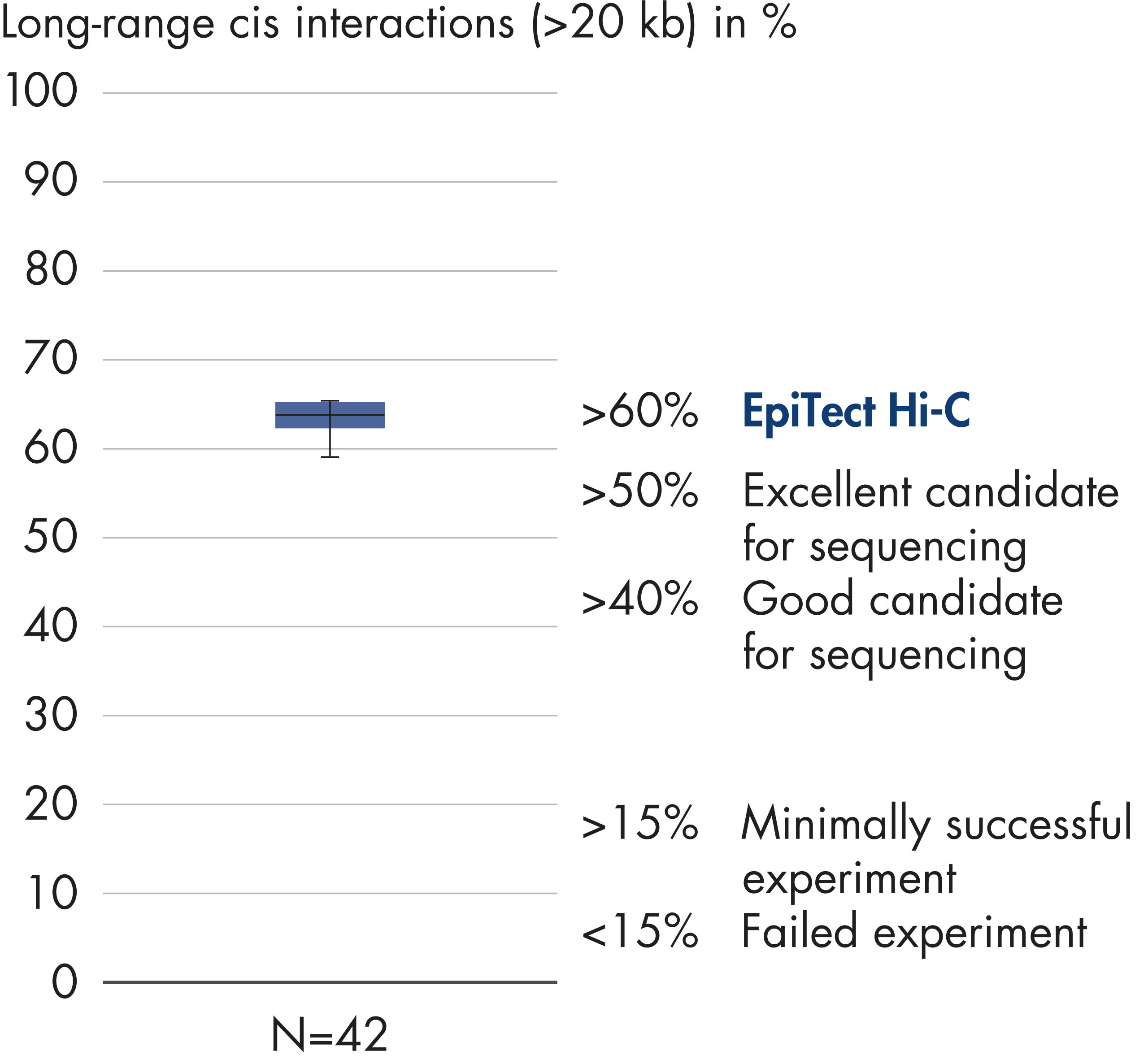
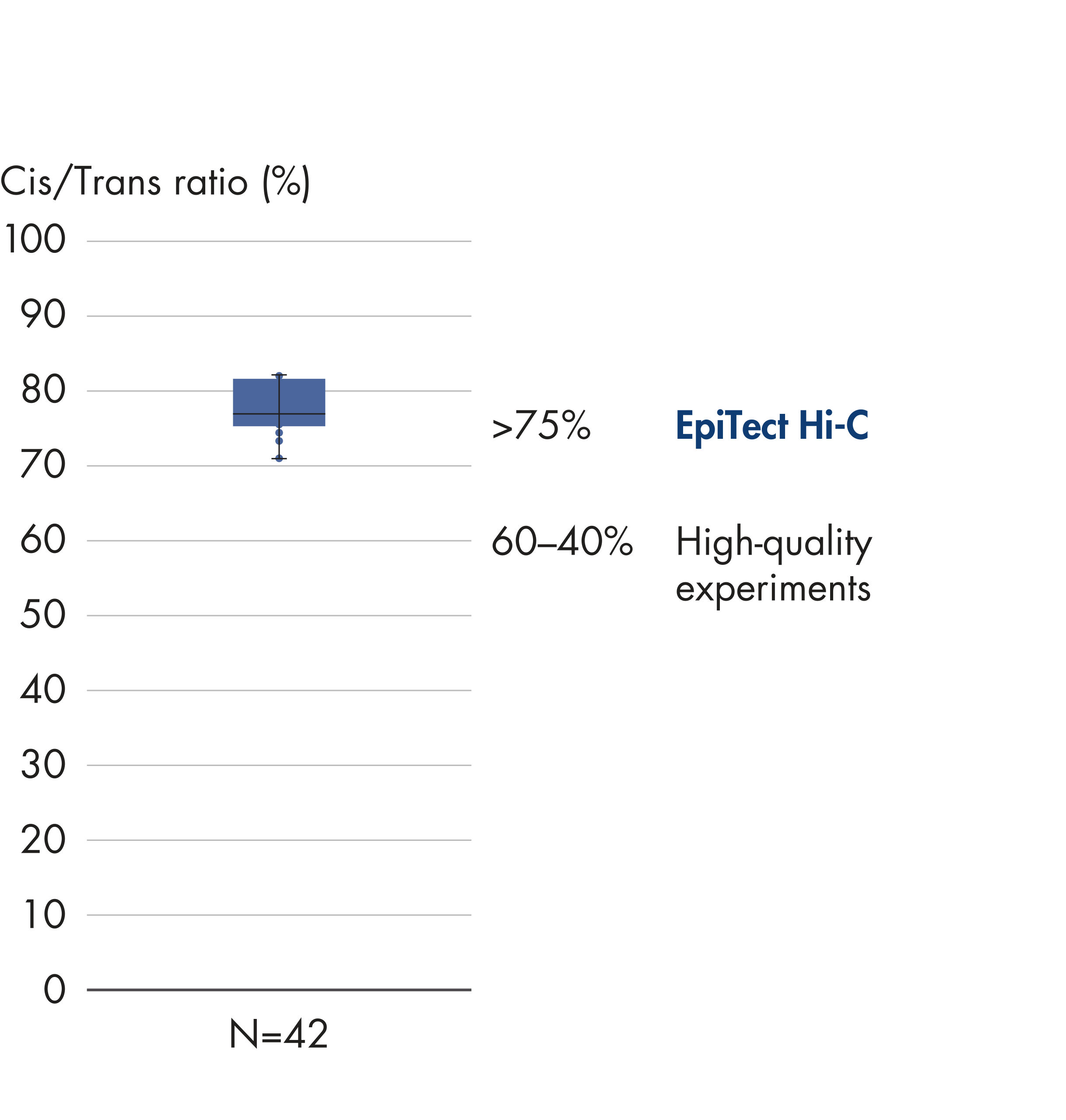
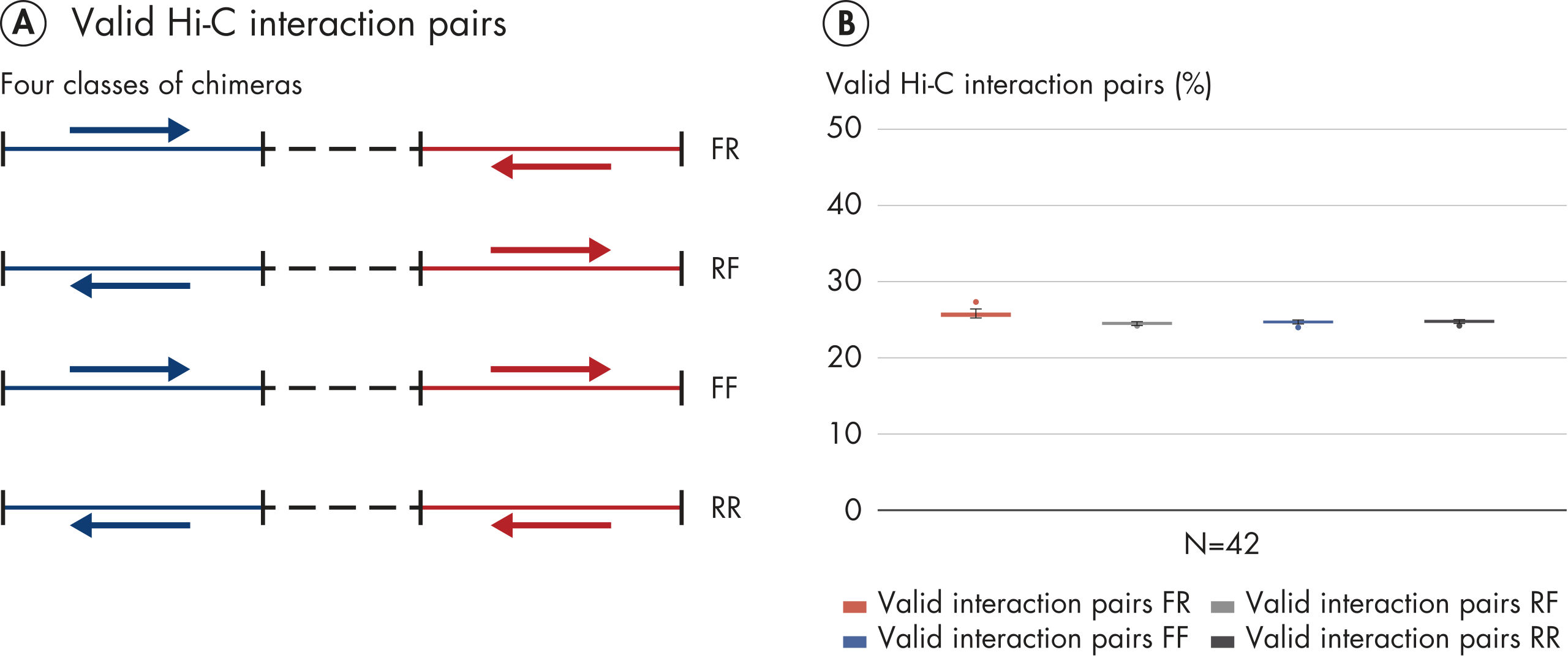
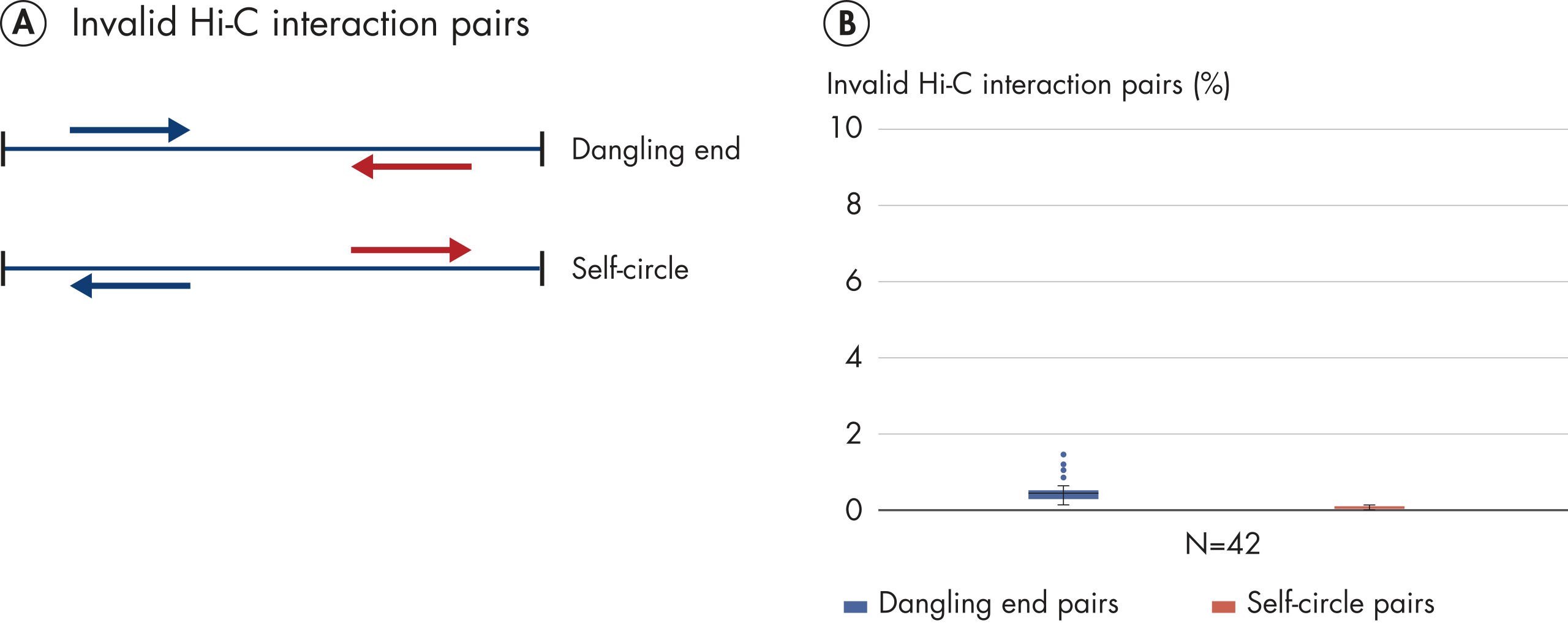
L’EpiTect Hi-C Kit produit des bibliothèques de NGS Hi-C de haute qualité, garantissant la génération de données de premier ordre d'un séquençage profond coûteux en aval. Pour évaluer les performances du kit, les résultats de séquençage de plus de 40 bibliothèques EpiTect Hi-C ont été analysés. Les mesures de CQ les plus importantes sont présentées dans les figures suivantes : Pourcentage d’événements Hi-C, Pourcentage d’interactions cis à longue portée, Rapport Cis/Trans, Pas de biais d’orientation des brins avec l’EpiTect Hi-C Kit et Pourcentage de lectures appariées provenant d’un seul fragment de restriction. Les données montrent que l’EpiTect Hi-C Kit produit des bibliothèques NGS qui, en moyenne, dépassent de loin les critères normalement considérés comme suffisants pour une expérience Hi-C réussie.
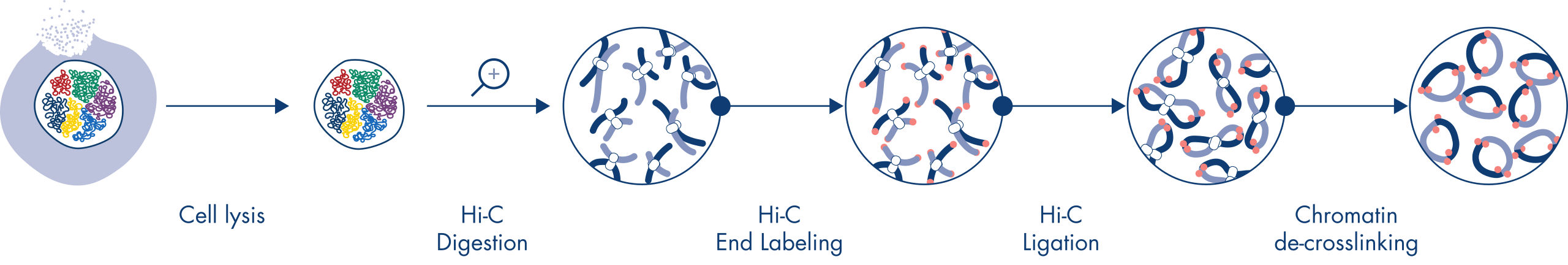
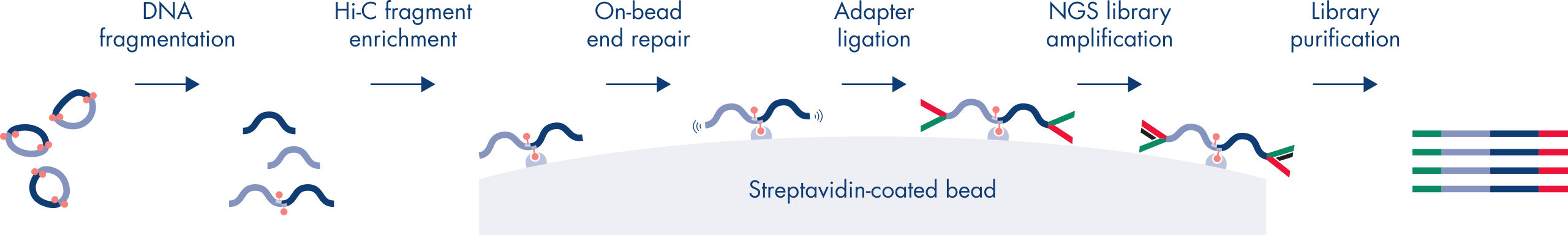
Hi-C est un dosage de ligature de proximité qui permet de capturer les interactions avec la chromatine à l’échelle du génome. L’EpiTect Hi-C Kit est une préparation d’ADN spécialisée qui permet d’obtenir une bibliothèque NGS compatible avec Illumina (voir figures Procédure EpiTect Hi-C – jour 1 et Procédure EpiTect Hi-C – jour 2). Le dosage commence par la purification de noyaux dans lesquels la conformation de la chromatine a été gelée par la réticulation chimique des protéines de liaison à l’ADN et de l’ADN. L’ADN est ensuite complètement digéré grâce à une enzyme de restriction de 4 pb. Les extrémités ouvertes de l’ADN sont marquées à la biotine puis ligaturées. Le séquençage en paires des produits de ligature Hi-C permet d’identifier un très grand nombre de séquences chimériques provenant de brins d’ADN qui étaient étroitement associés dans l’espace. La probabilité que deux séquences soient ligaturées ensemble est fonction de leur distance moyenne dans l’espace. La quantification des jonctions de ligature permet de déterminer les fréquences de contact avec l’ADN, à partir desquelles il est possible d’établir une cartographie à haute résolution du repliement de la chromatine.
La procédure EpiTect Hi-C (voir figures Procédure EpiTect Hi-C – jour 1 et Procédure EpiTect Hi-C – jour 2) représente une nette amélioration par rapport aux protocoles publiés. Une procédure compliquée d’une semaine a été transformée en un protocole simple et fiable qui ne nécessite qu’un jour et demi. En outre, le nombre d’échantillons requis a été réduit d’un ordre de grandeur, ce qui permet de créer des bibliothèques NGS Hi-C à partir de 5 000 cellules seulement. Le protocole a été développé pour travailler avec des cellules réticulées provenant de cultures de cellules de mammifères.
La procédure EpiTect Hi-C est une version de la méthode Hi-C in situ (c’est-à-dire dans le noyau) dans laquelle les noyaux sont délicatement purifiés et perméabilisés pour maintenir l’organisation spatiale du génome pendant les étapes initiales de digestion et de ligature. Ce processus est essentiel pour minimiser le bruit de fond provenant d’événements de ligature non informatifs qui ne reflètent pas l’organisation du génome. La raison en est que les noyaux intacts limitent les mouvements et les collisions aléatoires des complexes réticulés, de sorte que les événements de ligature se produisent principalement entre des fragments d’ADN topologiquement associés.
Construction de bibliothèques NGS Hi-C
La procédure d’utilisation de l’EpiTect Hi-C Kit se compose de 2 parties, chacune pouvant être réalisée en une journée. Les étapes du protocole sont résumées dans les tableaux ci-dessous et présentées dans les figures. Procédure EpiTect Hi-C – jour 1 et Procédure EpiTect Hi-C – jour 2. Les adaptateurs Illumina inclus sont dotés de codes-barres de séquence qui permettent le séquençage multiplex d’un maximum de 6 échantillons.
Pour consulter le protocole complet, voir le Manuel EpiTect Hi-C détaillé.
Analyse des données
L’analyse des données Hi-C est proposée dans notre GeneGlobe Data Analysis Center.. Les résultats du séquençage Hi-C peuvent être analysés à l’aide du portail d’analyse des données EpiTect Hi-C, qui fait appel à des outils open source pour fournir un rapport de séquençage CQ, des matrices de contact Hi-C et la visualisation des cartes de contact avec la chromatine. Pour plus d’informations, voir le EpiTect Hi-C Data Analysis Portal User Guide.
Conformation de la chromatine
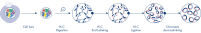
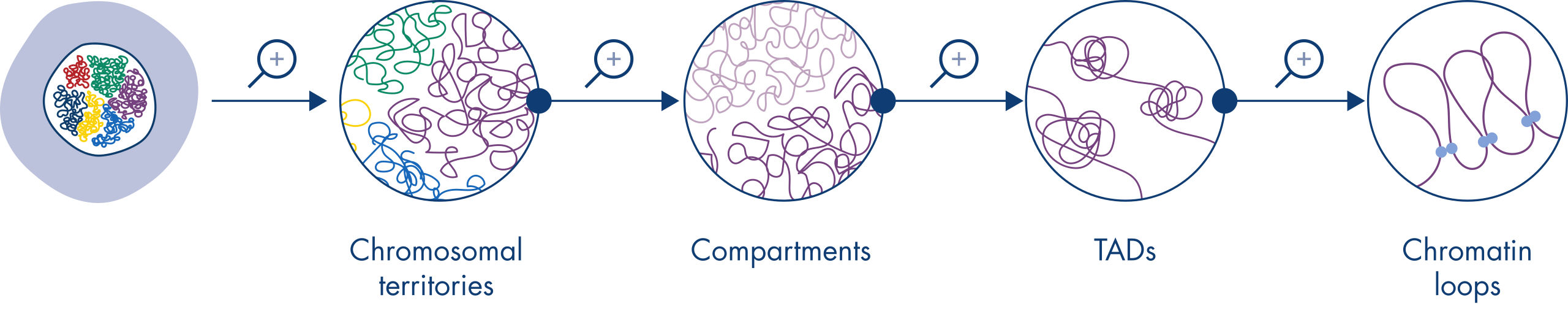
Hi-C est rapidement devenu un outil très important pour l’analyse de l’organisation nucléaire. L’analyse des données Hi-C a révélé une incroyable complexité de l’architecture du génome, avec de multiples couches d’organisation spatiale qui divisent le génome en territoires chromosomiques, sous-compartiments chromosomiques, domaines topologiquement associés (TAD) et boucles d’ADN à une résolution croissante (voir la figure Niveaux d’organisation de la chromatine). Par ailleurs, l’organisation du génome est dynamique et se modifie au cours du développement. Les chromosomes ne se plient pas de la même manière dans deux types de cellules.
Réarrangements chromosomiques et variations du nombre de copies
Les chromosomes individuels sont physiquement séparés en territoires distincts et, par conséquent, les interactions d’ADN capturées par Hi-C ont principalement lieu entre l’ADN du même chromosome (en cis) avec peu d’interactions entre les chromosomes (en trans). En raison de ce phénomène, il est possible d’utiliser Hi-C comme dosage à l’échelle du génome pour identifier les translocations et d’autres variantes structurelles d’intérêt. Par rapport à d’autres techniques NGS, Hi-C nécessite une couverture extrêmement faible, ce qui permet de réduire les coûts. En outre, ce type de réarrangement impliquant des régions peu cartographiables peut être mieux détecté avec Hi-C qu’avec les méthodes NGS standards. De façon pratique, les mêmes données Hi-C peuvent être utilisées pour détecter les modifications du nombre de copies.
Assemblage des génomes – Phasage des haplotypes
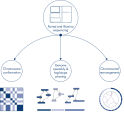
Lors du séquençage et de l’assemblage des génomes de nouvelles espèces, la génération d’échafaudages de séquences est souvent limitée par de grandes étendues de séquences répétitives qui s’étendent au-delà de la portée du séquençage. Dans les données Hi-C, la grande majorité des interactions se produisent en cis entre des loci situés sur le même chromosome. En outre, une part importante de ces interactions en cis est à longue portée, se produisant entre des loci séparés par des millions de bases d’ADN. Ces propriétés des interactions chromatiniennes peuvent être exploitées pour ordonner, orienter et assembler des échafaudages de séquences en chromosomes presque complets sans avoir besoin d’un génome de référence. Selon les mêmes principes, les cartes d’interaction Hi-C peuvent être employées pour créer des génomes diploïdes en attribuant des variants génétiques aux chromosomes frères paternels et maternels (voir figure Applications en aval des données de séquençage Hi-C).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}