
EpiTect Hi-C Kit
クロマチン構造の高分解能マッピング、ゲノム配列の高品質アセンブリー、ハプロタイプフェージングおよび染色体再配列の同定用。
クロマチン構造の高分解能マッピング、ゲノム配列の高品質アセンブリー、ハプロタイプフェージングおよび染色体再配列の同定用。
✓ オンライン注文による24時間年中無休の自動処理システム
✓ 知識豊富で専門的な製品&テクニカルサポート
✓ 迅速で信頼性の高い(再)注文
カタログ番号 / ID. 59971
✓ オンライン注文による24時間年中無休の自動処理システム
✓ 知識豊富で専門的な製品&テクニカルサポート
✓ 迅速で信頼性の高い(再)注文
Hi-Cはもともと、ゲノムワイドな染色体構造検出のための強力な技術として考案され、kbの分解能でクロマチン構造の特性解析を可能にしました。しかし、この技術は他にも重要なアプリケーションがあります。たとえば、Hi-Cは、既知の参照ゲノムを持たない生物から、少数の非常に長いスキャフォールドを持つ連続性の高いゲノムアセンブリーを作成するために使用されます。さらに、Hi-Cはハプロタイプフェージングや染色体再配列の検出にも非常に有用です。
EpiTect Hi-C Kitは、低細胞インプット要件の、堅牢かつシンプルで迅速なプロトコールを提供し、クロスリンクされた細胞から高品質のHi-C Illumina NGSライブラリーを2日かけずに作成することができます。
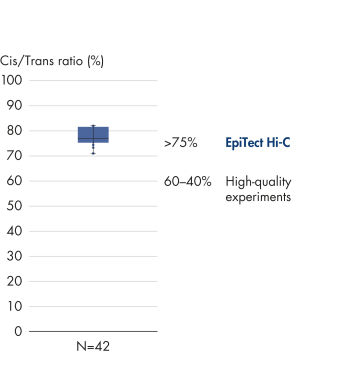
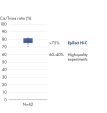
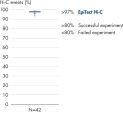
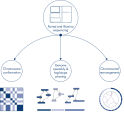
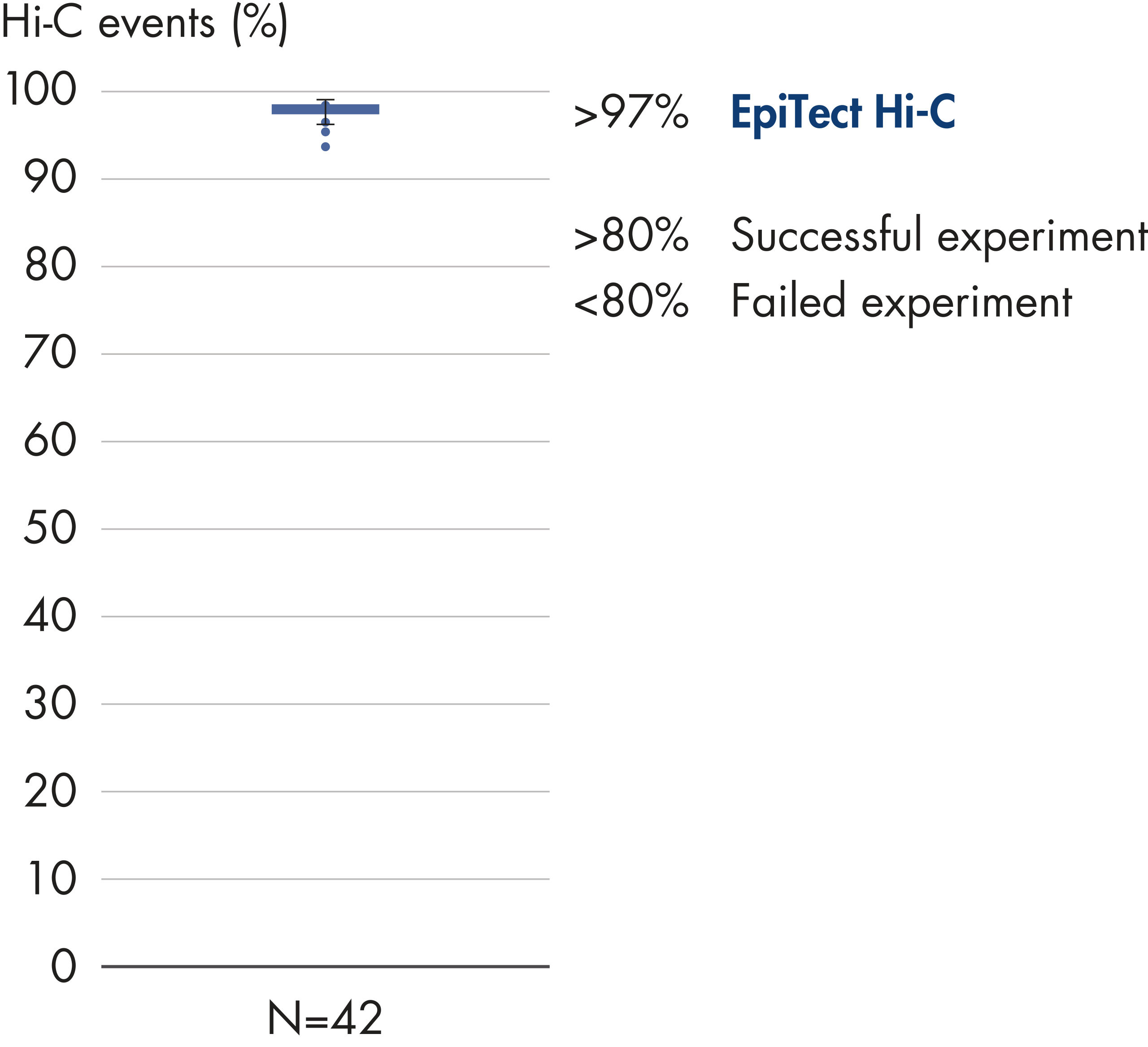
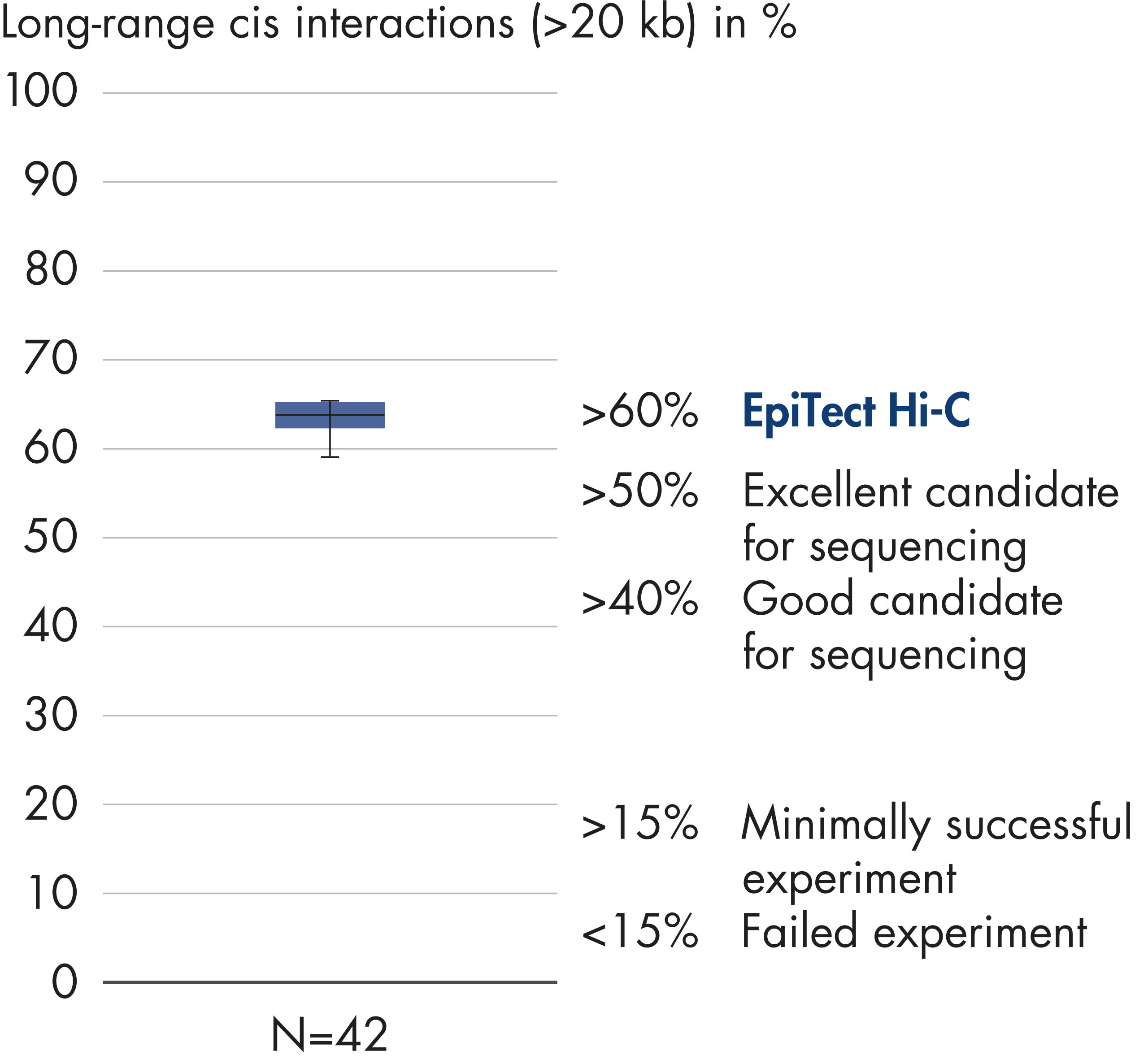
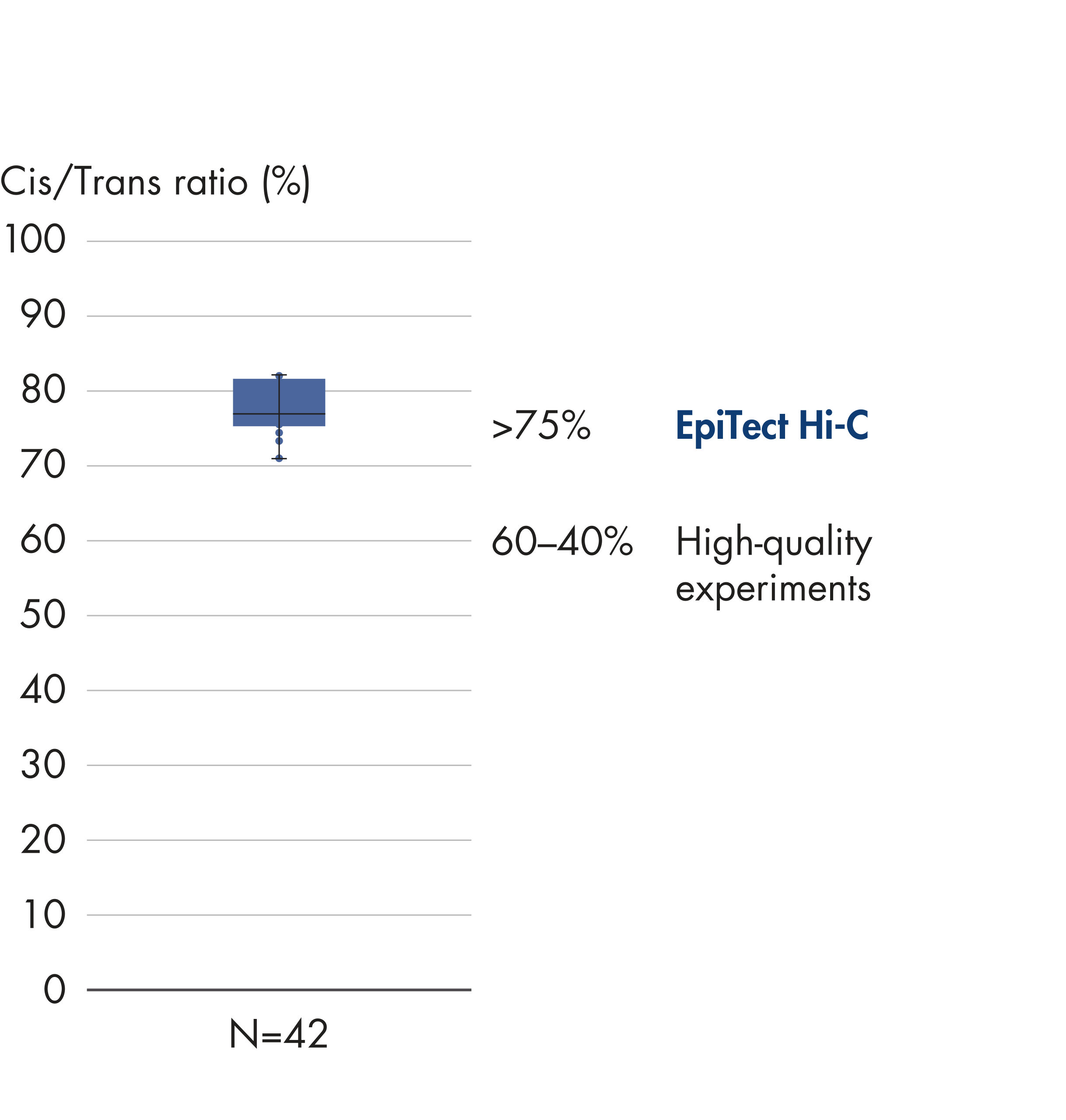
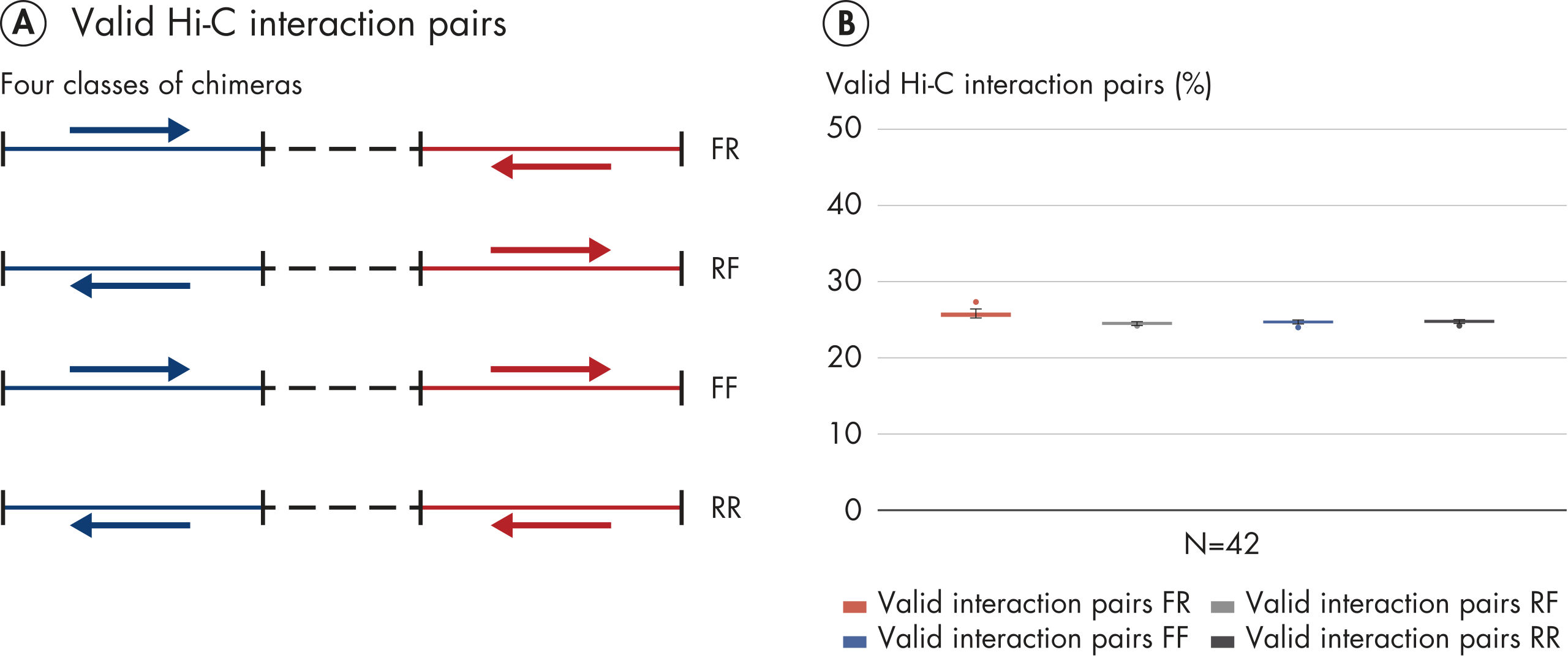
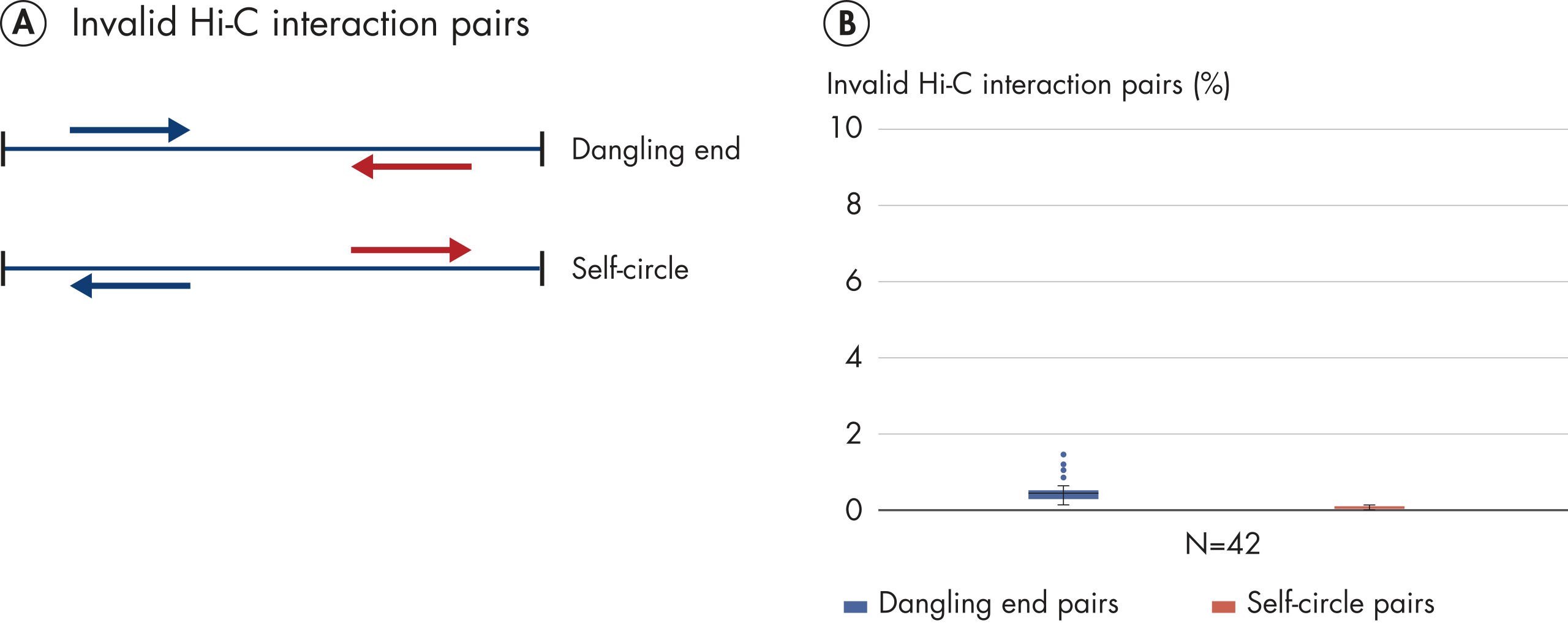
EpiTect Hi-C Kitは高品質のHi-C NGSライブラリーを作成することができることによって、コストのかかるダウンストリームのディープシークエンシングを行うことなく最高品質のデータを確実に生成します。40を超えるEpiTect Hi-Cライブラリーのシークエンシング結果を分析し、キットの性能を評価しました。最も重要なQCメトリクスを Hi-Cイベントの割合(%)、 長距離シス相互作用の割合(%)、 シス/トランス比、 鎖配向バイアスのないEpiTect Hi-C Kit、および 単一の制限フラグメント由来のペアリードの割合(%)の図に示します。データは、EpiTect Hi-C Kitが、平均してHi-C実験の成功に通常十分と考えられる基準をはるかに上回るNGSライブラリを生成することを示しています。
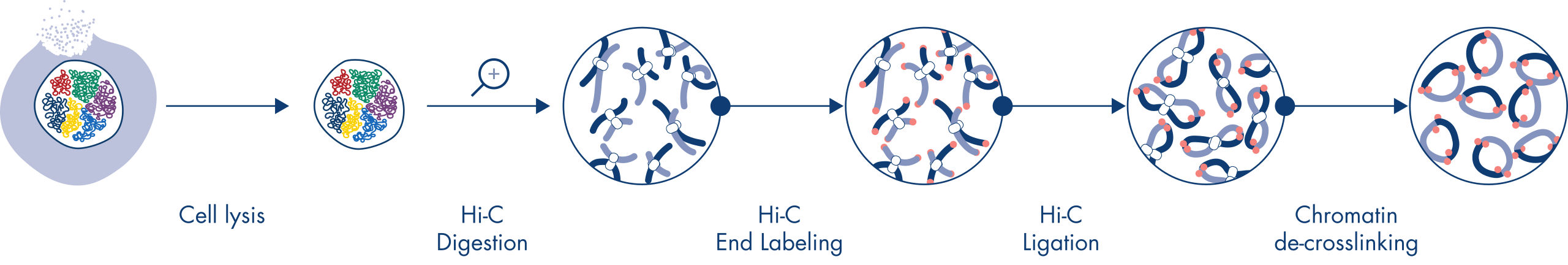
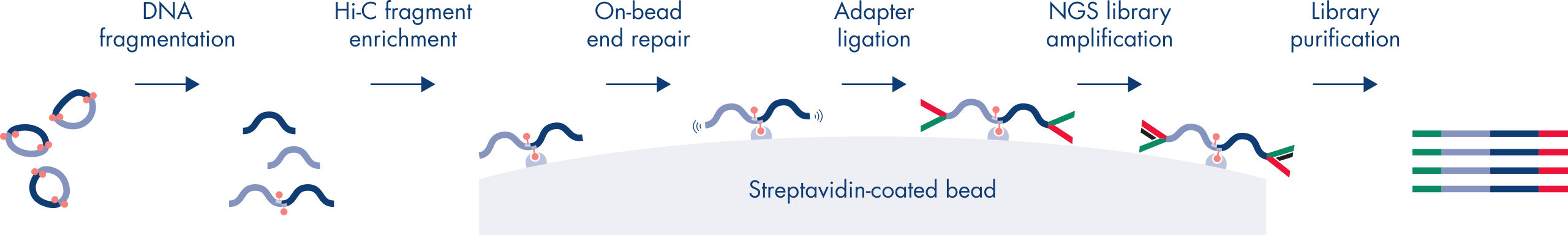
Hi-Cは、ゲノムワイド規模でクロマチン相互作用を捉える近接ライゲーションアッセイです。EpiTect Hi-C Kitでは、特殊なDNA調製により、Illuminaに対応するNGSライブラリが得られます(図 EpiTect Hi-C ワークフロー – 1日目および EpiTect Hi-Cワークフロー – 2日目参照)。簡単に説明しますと、このアッセイは、DNA結合タンパク質とDNAの化学的クロスリンクによってクロマチンコンフォメーションが固定されている核の精製から始まります。次に、DNAは、4bpの制限酵素で完全に消化されます。開いたDNA末端をビオチンで標識し、その後ライゲーションを行います。Hi-Cライゲーション産物のペアエンドシークエンシングにより、空間的に密接に関連しているDNA鎖に由来する非常に多くのキメラ配列を特定します。2つの配列が一緒にライゲーションされる確率は、空間における両者の平均距離の関数です。ライゲーションジャンクションを定量化すると、DNA接触頻度を決定することができ、そこからクロマチン構造の高分解能マッピングを行うことができます。
EpiTect Hi-Cワークフロー(図 EpiTect Hi-Cワークフロー – 1日目と EpiTect Hi-Cワークフロー – 2日目参照)は、公開されているプロトコールに比べ、大幅に改善されています。1週間かかる複雑な手順が、わずか1.5日で完了するシンプルで堅牢なプロトコールに変わりました。さらに、サンプルインプット要件は1桁減少し、わずか5000個の細胞からHi-C NGSライブラリを作成できます。このプロトコールは、哺乳類細胞培養からサンプリングしたクロスリンクされた細胞を基に開発されました。
EpiTect Hi-C手順は、in situ (核内) Hi-C法のひとつのバージョンで、核を穏やかに精製し、透過処理を行います。最初の消化とライゲーションステップにおいても、ゲノムの空間的構成を維持します。このプロセスは、ゲノムの構成を反映しない、情報価値のないライゲーションイベントによるバックグラウンドノイズを最小限に抑えるために不可欠です。これは、インタクトな核が架橋複合体の動きやランダムな衝突を抑制し、ライゲーションイベントが主に位相的に関連するDNA断片間で発生するからです。
Hi-C NGSライブラリの構築
EpiTect Hi-C Kitのワークフローは2つの部分で構成されており、それぞれ1日で完了します。プロトコールの手順を以下の表にまとめ、図 EpiTect Hi-C ワークフロー – 1日目と EpiTect Hi-Cワークフロー – 2日目で可視化します。付属のIlluminaアダプターには配列バーコードがあり、最大6つのサンプルのマルチプレックスシークエンシングが可能です。
全体のプロトコールをご覧になるには、当社の詳細なEpiTect Hi-Cハンドブックをご覧ください。
データ分析
Hi-Cデータ分析は、当社のGeneGlobeデータ分析センターより提供しています。Hi-Cシークエンシング結果は、EpiTect Hi-C Data Analysis Portalを使用して分析できます。このポータルはオープンソースツールを使用し、QCシークエンシングレポート、Hi-Cコンタクトマトリックス、クロマチンコンタクトマップを可視化します。詳細については、当社のEpiTect Hi-Cデータ分析ポータルユーザーガイドをご覧ください。
クロマチン構造
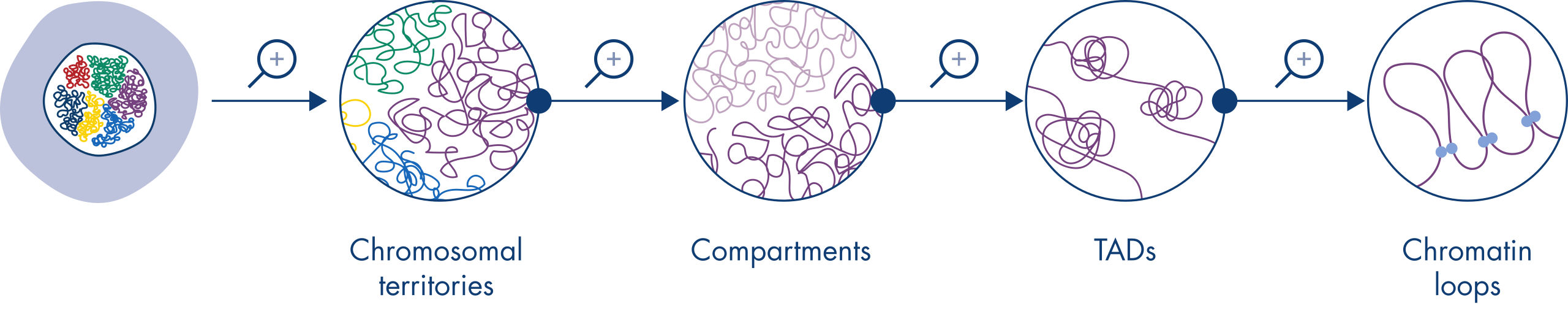
Hi-Cは、核組織分析のための非常に重要なツールになりました。Hi-Cデータの分析により、ゲノムの構造が驚くほど複雑であることが明らかになりました。解像度を上げると、ゲノムを染色体テリトリー、染色体サブコンパートメント、トポロジー関連ドメイン(TADs)、DNAループに分割できる多層構造になっていることがわかります(図 クロマチン構造のレベル参照)。さらに、ゲノムの構造はダイナミックで、発生過程で変化します。染色体が同じように折り畳まれている細胞タイプは2つとありません。
染色体再配列とコピー数多型
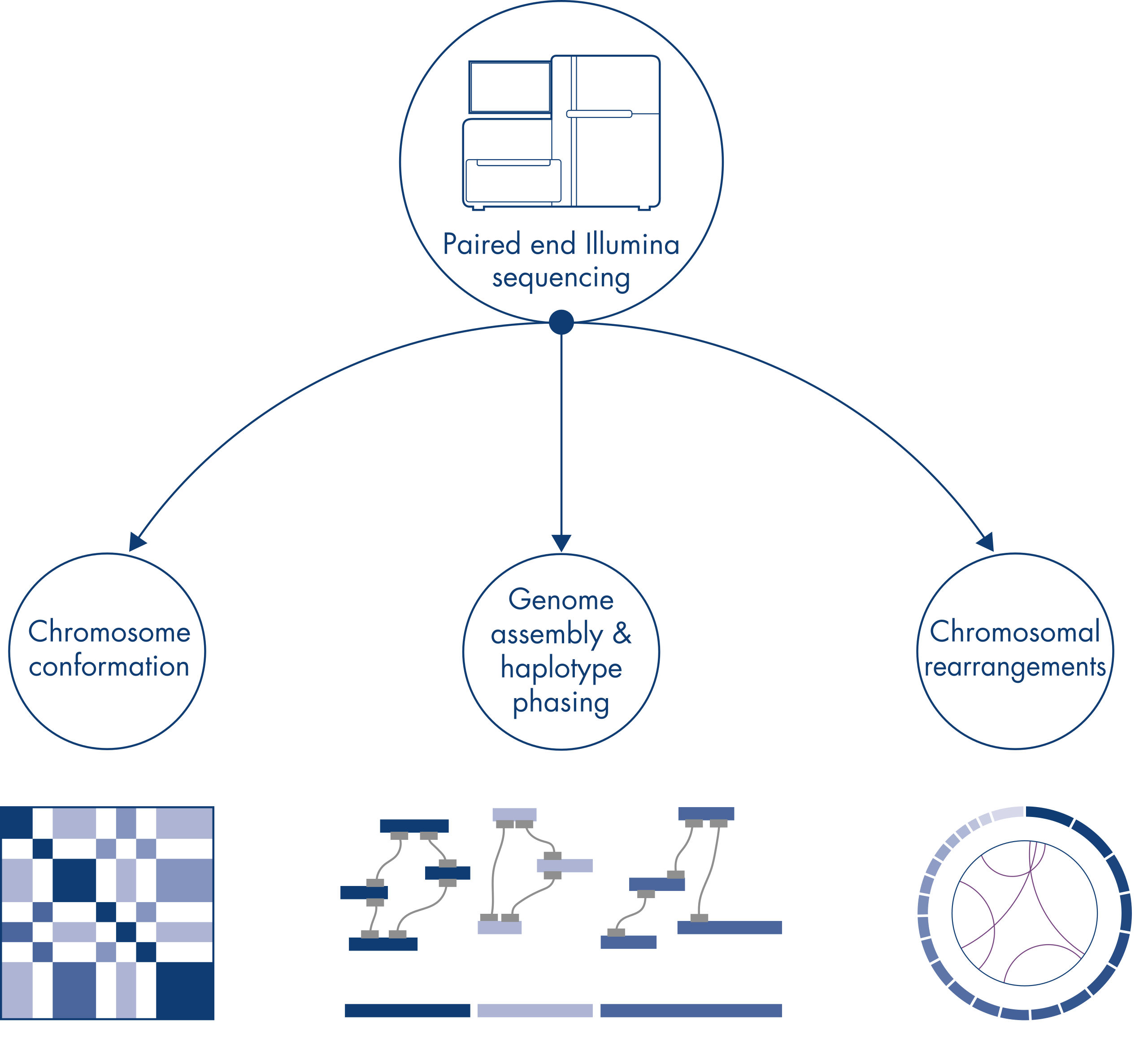
個々の染色体は物理的に個別のテリトリーに分かれているため、Hi-Cが捉えるDNA相互作用は、主として同じ染色体のDNA間(シス)で起こり、染色体間(トランス)ではほとんど起こりません。この現象により、Hi-Cは転座やその他の対象構造変異を特定するためのゲノムワイドアッセイとして使用できます。他のNGS技術と比較して、Hi-Cは非常に低いカバレッジしか必要としないため、コストを節約できます。さらに、マッピングが不十分な領域が関与する再配列は、標準的なNGS法よりもHi-Cを用いた方が検出しやすくなります。便利なことに、同じHi-Cデータを用いてコピー数の変化を検出できます。
ゲノムアセンブリ – ハプロタイプフェージング
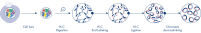
新しい種のゲノムのシークエンシングやアセンブリを行うとき、配列スキャフォールドの作成は、シークエンシング範囲を超える長い反復配列によって制限されることがよくあります。Hi-Cデータでは、相互作用の大部分が同じ染色体上の遺伝子座間のシスで生じます。さらに、このシス相互作用の大部分は長距離であり、数百万ものDNA塩基によって隔てられた遺伝子座間で生じます。このようなクロマチン相互作用の特性を利用することで、参照ゲノムを必要とせずに、配列スキャフォールドを順序付け、方向付け、結合して、ほぼ全長の染色体にすることができます。同じ原理を用いて、Hi-C相互作用マップを使用し、遺伝子変異を父方および母方の姉妹染色体に割り当てることによって、二倍体ゲノムを作成できます(図 Hi-Cシークエンシングデータのダウンストリームアプリケーション参照)。





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}